美吉生物结题报告
原核转录组云分析
客户: 宋晓宁
2023年12月28日
目录
一、项目信息
| 项目名称 | |||
|---|---|---|---|
| 原核转录组云分析 | |||
| 合同编号 | |||
| MJ20231128302 | |||
| 项目样本信息 | |||
| 样本来源 | 乳酸菌_保加利亚乳杆菌 | ||
| 样本类型 | 菌体沉淀 | ||
| 样本数量 | 4 | ||
| 备注 | Novaseq | ||
| 客户信息 | |||
| 单位名称 | 中国海洋大学 | ||
| 单位地址 | 山东省青岛市 | ||
| 项目联系人 | 宋晓宁 | 电话 | 15689130301 |
| 邮箱 | songxn0126@163.com | ||
| 美吉联系人信息 | |||
| 销售员 | 王嘉玮 | 电话 | 18443147121 |
| 邮箱 | jiawei.wang@majorbio.com | ||
| 技术支持 | 赵海霞1 | 电话 | 021-20725162 |
| 邮箱 | meta@majorbio.com | ||
二、工作流程
实验采用TruSeqTM Stranded Total RNA Library Prep Kit试剂构建文库,在合成cDNA第二条链的dNTPs试剂中用dUTP代替dTTP,使cDNA第二链中碱基包含A/U/C/G。在PCR扩增前,用UNG酶将cDNA第二链消化,从而使文库中仅包含cDNA第一链。
2.1 提取total RNA
从组织样品中提取total RNA,利用Nanodrop2000对所提RNA的浓度和纯度进行检测,琼脂糖凝胶电泳检测RNA完整性,Agilent2100测定RIN值。单次建库要求RNA总量2ug,浓度≥ 100ng/μL,OD260/280介于1.8~2.2之间。
2.2 去除rRNA
不同于真核生物mRNA 3‘末端具有ployA尾,原核生物不能利用Oligo dT与ployA进行A-T碱基配对,从总RNA中分离出mRNA。一般采用去除rRNA的方式,用于分析转录组信息。
2.3 片段化mRNA
去除rRNA后,对富集得到的mRNA是完整的RNA序列,平均长度达几kb,因此需要对其进行随机打断。加入fragmentation buffer,可以将mRNA随机断裂成200bp左右的小片段。
2.4 反转合成cDNA
在逆转录酶的作用下,利用随机引物,以 mRNA 为模板反转合成一链 cDNA,进行二链合成时,dNTPs 试剂中用 dUTP代替 dTTP,使 cDNA 第二链中碱基包含 A/U/C/G。
2.5 连接adaptor
双链的 cDNA 结构为粘性末端,加入 End Repair Mix 将其补成平末端,随后在 3’末端加上一个 A 碱基,用于连接 Y 字形的接头。
2.6 UNG酶消化cDNA二链
在 PCR 扩增前,用 UNG 酶将 cDNA 第二链消化,从而使文库中仅包含 cDNA 第一链。
2.7 上机测序
2.7.1 NovaSeqXPlus平台上机测序
①Qubit 4.0定量,按数据比例混合上机;
②cBot上进行桥式PCR扩增,生成clusters;
③上机测序。
2.7.2 DNBSEQ-T7 平台上机测序
①文库DNA单链环化;
②去除未环化序列,并进行纯化;
③制备DNA纳米球(DNA nanoball, DNB);
④上机测序。
注:用户可根据选择的高通量二代测序平台参考上述上机测序流程。
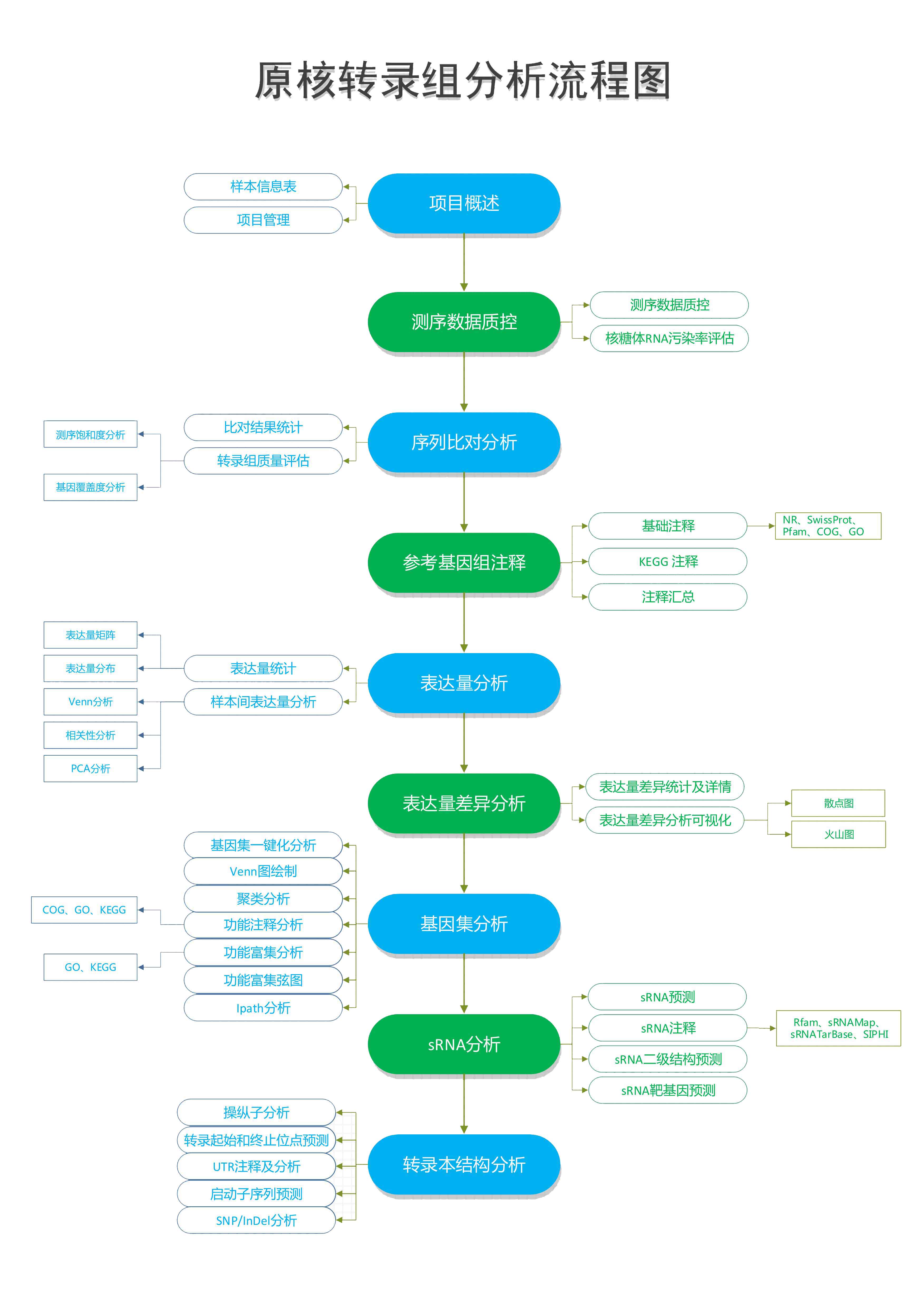
三、分析报告
3.1 测序数据质控
为方便测序数据的分析、 发布和共享, 测序平台通过将测序图像信号经CASAVA碱基识别( Base Calling) 转换成文字信号, 并将其以 fastq格式储存起来作为原始数据。 根据index序列区分各个样本的数据, 以便进行后续分析。 在fastq文件中每条序列 由4行数据组成, 其中第一行和第三行为读段识别码( 第一行以“@”开头, 第三行以“+”开头) , 第二行为碱基序列, 而第四行是第二行序列的各碱基所对应的测序质量值。
高通量二代测序仪单次运行能产生数十亿级的reads, 如此海量的数据无法逐个展示每条read的质量情况; 因此我们运用统计学的方法, 对所测序列进行统计和质控, 可以从宏观上直观地反映出样本的文库构建质量和测序质量。
测序数据质控共包括三部分内容: 1. 测序数据统计; 2. 原始数据统计; 3. 质控数据统计。
3.1.1 测序数据质控
运用统计学的方法对所有测序reads的每个cycle进行碱基分布和质量波动的统计,可以从宏观上直观地反映出测序样本的文库构建质量和测序质量,并对每一个样本的碱基质量、碱基错误率以及碱基分布进行分析。
Raw Reads碱基错误率分布图

注:图为质控前碱基错误率分布图。横坐标是reads 碱基坐位置,表示由5’到3’端在reads上的碱基排列顺序;纵坐标是所有reads 在该位点处碱基的平均错误率(%)。前半部分为双端测序序列的第一端测序Reads的错误率分布情况,后半部分为另一端测序reads 的错误率分布情况。横坐标为reads 的碱基位置,纵坐标为单碱基错误率。
Clean Reads碱基组成分布图

注:质控后碱基组成分布图。横坐标是reads 碱基坐标,表示由5’到3’端在reads上的碱基排列顺序;纵坐标是所有reads 在该测序位置A、C、G、T、N 碱基分别占的百分比,不同碱基用不同颜色表示。序列的起始位置与测序的引物接头相连,因此A、C、G、T 在起始端会有所波动,后面会趋于稳定。模糊碱基N 所占比例越低,说明未知碱基越少,测序样本受系统AT 偏好影响越小。
Raw Reads碱基组成分布图

注:质控前碱基组成分布图。横坐标是reads 碱基坐标,表示由5’到3’端在reads上的碱基排列顺序;纵坐标是所有reads 在该测序位置A、C、G、T、N 碱基分别占的百分比,不同碱基用不同颜色表示。序列的起始位置与测序的引物接头相连,因此A、C、G、T 在起始端会有所波动,后面会趋于稳定。模糊碱基N 所占比例越低,说明未知碱基越少,测序样本受系统AT 偏好影响越小。
Clean Reads碱基质量分布图

注:图为质控后碱基质量分布图。横坐标是reads 碱基座位置,表示由5’到3’端在reads上的碱基排列顺序;纵坐标是所有reads 在该位点处碱基的Q值。前半部分为双端测序序列的第一端测序Reads的Q值分布情况,后半部分为另一端测序reads 的Q值分布情况。横坐标为reads 的碱基位置,纵坐标为Q值。
Clean Reads碱基错误率分布图

注:图为质控后碱基错误率分布图。横坐标是reads 碱基坐位置,表示由5’到3’端在reads上的碱基排列顺序;纵坐标是所有reads 在该位点处碱基的平均错误率(%)。前半部分为双端测序序列的第一端测序Reads的错误率分布情况,后半部分为另一端测序reads 的错误率分布情况。横坐标为reads 的碱基位置,纵坐标为单碱基错误率。
Raw Reads碱基质量分布图

注:图为质控前碱基质量分布图。横坐标是reads 碱基座位置,表示由5’到3’端在reads上的碱基排列顺序;纵坐标是所有reads 在该位点处碱基的Q值。前半部分为双端测序序列的第一端测序Reads的Q值分布情况,后半部分为另一端测序reads 的Q值分布情况。横坐标为reads 的碱基位置,纵坐标为Q值。
3.2 序列比对分析
将质控后的原始数据,即clean data(reads),与参考基因组进行比对,获得用于后续分析的 mapped data(reads),同时对本次转录组测序的比对结果进行质量评估,主要包括测序饱和度、基因覆盖度、Reads在参考基因组不同区域分布以及Reads在不同染色体分布分析。
3.2.1 转录组质量评估
对本次测序的比对结果进行整体质量评估, 展示测序饱和度、 测序覆盖度、不同区域Reads分布以及不同染色体Reads分布情况。
3.2.1.1 测序饱和度分析
测序饱和度曲线图

注:横坐标为有效比对reads的百分比 (如:60表示随机抽取60%的比对测序数据用于计算该基因的表达量)。纵轴为该取样条件下表达量与终值的偏差比例(误差15%以内) (如:0.4表示有40% 的基因表达量误差在15%以内),数值越趋近于1 则表示表达量越趋于饱和。每种颜色线条代表该样品中不同表达水平的基因表达量饱和度曲线。通常大部分中等以上表达量的基因(即表达量值在3.5以上的基因)在测序reads的40%比对上时接近饱和(纵轴数值趋近于1),则说明饱和度总体质量较高,该测序量能够覆盖绝大多数的表达基因。
3.2.1.2 测序覆盖度分析
测序覆盖度分布图

注:横坐标为单个基因的碱基长度占总碱基长度的百分比,0表示基因的5’端,100表示基因的3’端;纵坐标为比对到所有基因的横轴位置上相应区间内的序列条数的总和。图中体现了所有基因覆盖情况的叠加结果,曲线中每个点的纵坐标表示所有基因在该相对比例位置上所有序列的数量;曲线反映了测序所得序列是否在基因上均匀分布。若无明显偏向锋,则说明测序无偏向性。
3.3 功能注释与查询
3.3.1 基础注释
将参考基因组与NR、Swiss-Prot、Pfam、COG、GO、KEGG等数据库进行比对,全面获得基因的功能信息并对各数据库注释情况进行统计。
3.3.1.1 基础注释统计
将基因与NR,Swiss-Prot,Pfam,COG,GO和KEGG等数据库比对的整体情况进行统计。包括基础注释统计Venn图,基础注释统计柱状图,基础注释统计结果表。
基础注释统计Venn图

注:不同颜色的代表不同数据库的注释结果,其中的数值代表注释到某一数据库的基因数目。共有区域代表同时注释到多个数据库的基因数目,特有区域代表仅注释到该数据库的基因数目。
基础注释统计柱状图

注:横坐标表示不同的数据库,具体可见右上角图例;纵坐标表示不同的数据库注释到的基因的数目。
基础注释统计结果表
| Genome Size (bp) | GC Content(%) | CDS No. | Genes of NR (Percent(%)) | Genes of Swiss-Prot (Percent(%)) | Genes of Pfam (Percent(%)) | Genes of COG (Percent(%)) | Genes of GO (Percent(%)) | Genes of KEGG (Percent(%)) |
|---|---|---|---|---|---|---|---|---|
| 1865847 | 49.75 | 2051 | 2013(98.15) | 1361(66.36) | 1578(76.94) | 1600(78.01) | 1517(73.96) | 1100(53.63) |
注:(1)Genome Size (bp):基因组大小;(2)GC Content(%):GC含量;(3)CDS No.:编码基因数目;(4)Genes of NR (Percent(%)):注释到NR数据库的基因的数目及占总基因数的百分比;(5)Genes of Swiss-Prot (Percent(%)):注释到Swiss-Prot数据库的基因的数目及占总基因数的百分比;(6)Genes of Pfam (Percent(%)):注释到Pfam数据库的基因的数目及占总基因数的百分比;(7)Genes of COG (Percent(%)):注释到COG数据库的基因的数目及占总基因数的百分比;(8)Genes of GO (Percent(%)):注释到GO数据库的基因的数目及占总基因数的百分比;(9)Genes of KEGG (Percent(%)):注释到KEGG数据库的基因的数目及占总基因数的百分比;(Organism和Genome ID代表物种名称和基因组编号:具有明确的NCBI来源时显示)
3.3.1.2 COG注释
COG 是Clusters of Orthologous Groups of proteins的缩写(http://www.ncbi.nlm.nih.gov/COG/)。COG是在对已完成基因组测序的物种的蛋白质序列进行相互比较的基础上构建的,COG数据库选取的物种包括各个主要的系统进化谱系。每个COG家族至少由来自3个系统进化谱系的物种的蛋白所组成,所以一个COG对应于一个古老的保守结构域。构成每个COG的蛋白被假定来自于同一个祖先蛋白。进行COG数据库比对可以对预测蛋白进行功能注释、归类以及蛋白进化分析。
COG分类统计柱状图

注:横坐标代表COG的功能分类(用大写字母A~Z表示,具体含义请见COG注释结果详情表),纵坐标表示具有该类功能的基因数目。
3.3.1.3 GO注释
GO是基因本体论Gene Ontology的缩写(详情请见:http://www.geneontology.org/)。由于不同物种、不同数据库中的关于基因和基因产物等生物学术语的描述存在差异,当查询某个研究领域的相关信息时,生物学家需要花费大量的时间和精力去分析生物学术语之间的联系,而Gene
Ontology项目的目的就是为了标准化这些生物学术语,方便生物学家之间的相互交流。GO注释包括3个方面的内容:
Cellular component:the parts of a cell or its extracellular environment;
Molecular function:the elemental activities of a gene product at the molecular level, such as binding or catalysis;
Biological process:operations or sets of molecular events with a
defined beginning and end, pertinent to the functioning of integrated
living units: cells, tissues, organs, and organisms.
因此,GO注释更加便于我们理解基因背后所代表的生物学意义。
GO注释结果统计表
| GO Category No. | Gene No. of CC | Gene No. of MF | Gene No. of BP | Gene No. | Percent of All Genes (%) |
|---|---|---|---|---|---|
| 3 | 809 | 1247 | 827 | 1517 | 73.963920039005 |
注:(1)GO Category No.:GO功能分类数量;(2)Gene No. of Cellular Component:与细胞组成相关的基因数量;(3)Gene No. of Molecular Function:与分子功能相关的基因数量;(4)Gene No. of Biological Process:与生物过程相关的基因数量;(5)Gene No.:注释到GO的基因数量;(6)Percent of All Genes:注释到GO的基因占所有基因的百分比。
GO注释结果详情表
| Category | Function Description | GO ID (Level2) | Gene No. |
|---|---|---|---|
| molecular_function | catalytic activity | GO:0003824 | 891 |
| cellular_component | cellular anatomical entity | GO:0110165 | 753 |
| molecular_function | binding | GO:0005488 | 715 |
| biological_process | cellular process | GO:0009987 | 658 |
| biological_process | metabolic process | GO:0008152 | 658 |
| molecular_function | transporter activity | GO:0005215 | 168 |
| cellular_component | protein-containing complex | GO:0032991 | 121 |
| molecular_function | ATP-dependent activity | GO:0140657 | 90 |
| biological_process | localization | GO:0051179 | 85 |
| biological_process | biological regulation | GO:0065007 | 81 |
注:(1)Category:GO分类;(2)Function Description:功能描述;(3)GO ID:GO的ID编号;(4)Gene No.:基因数量。
GO注释分类统计柱状图

注:横坐标代表GO的三大分支即BP(Biological Process,生物过程)、CC(Cellular Component,细胞组分)、MF(Molecular Function,分子功能),以及更进一步的leve2、leve3或leve4分类(可进行切换展示);纵坐标代表该功能类别下的基因数目。
GO注释分类统计柱状图

注:横坐标代表GO的三大分支即BP(Biological Process,生物过程)、CC(Cellular Component,细胞组分)、MF(Molecular Function,分子功能),以及更进一步的leve2、leve3或leve4分类(可进行切换展示);纵坐标代表该功能类别下的基因数目。
GO注释分类统计饼图

注:每个饼图代表GO的不同分支即BP(Biological Process,生物过程)、CC(Cellular Component,细胞组分)、MF(Molecular Function,分子功能)。饼图的不同颜色代表GO level2、leve3或leve4分类(可进行切换展示),其面积表示该分类下注释到的基因数目。
3.3.1.4 KEGG 注释
在生物体内,基因产物并不是孤立存在而各自发挥作用的,不同基因产物之间通过有序的相互协调来一起行使具体的生物学功能。因此,KEGG数据库中丰富的通路信息将有助于我们从系统水平去了解基因的生物学功能,例如代谢途径、遗传信息传递以及细胞学过程等一些复杂的生物过程。
Pathway分类统计柱状图

注:纵坐标为KEGG代谢通路的名称,横坐标为注释到该通路的基因数目。KEGG代谢通路可分为7大类:代谢(Metabolism),遗传信息处理(Genetic Information Processing),环境信息处理(Environmental Information Processing),细胞过程(Cellular Processes),生物体系统(Organismal Systems),人类疾病(Human Diseases),药物开发(Drug Development)。(注:根据具体物种情况,获得的分类数目不同)。
Pathway信号通路注释结果统计表
| First Category | Second Category | Gene No. |
|---|---|---|
| Metabolism | Amino acid metabolism | 90 |
| Metabolism | Biosynthesis of other secondary metabolites | 23 |
| Metabolism | Carbohydrate metabolism | 91 |
| Metabolism | Energy metabolism | 57 |
| Metabolism | Global and overview maps | 49 |
| Metabolism | Glycan biosynthesis and metabolism | 47 |
| Metabolism | Lipid metabolism | 35 |
| Metabolism | Metabolism of cofactors and vitamins | 56 |
| Metabolism | Metabolism of other amino acids | 27 |
| Metabolism | Metabolism of terpenoids and polyketides | 14 |
| Metabolism | Nucleotide metabolism | 71 |
| Metabolism | Xenobiotics biodegradation and metabolism | 14 |
| Genetic Information Processing | Folding, sorting and degradation | 29 |
| Genetic Information Processing | Replication and repair | 47 |
| Genetic Information Processing | Transcription | 5 |
| Genetic Information Processing | Translation | 75 |
| Environmental Information Processing | Membrane transport | 111 |
| Environmental Information Processing | Signal transduction | 25 |
| Cellular Processes | Cell growth and death | 12 |
| Cellular Processes | Cell motility | 4 |
| Cellular Processes | Cellular community - prokaryotes | 52 |
| Cellular Processes | Transport and catabolism | 2 |
| Organismal Systems | Aging | 4 |
| Organismal Systems | Digestive system | 3 |
| Organismal Systems | Endocrine system | 9 |
| Organismal Systems | Environmental adaptation | 4 |
| Organismal Systems | Immune system | 2 |
| Organismal Systems | Nervous system | 1 |
| Human Diseases | Cancer: overview | 9 |
| Human Diseases | Cancer: specific types | 1 |
| Human Diseases | Cardiovascular disease | 7 |
| Human Diseases | Drug resistance: antimicrobial | 35 |
| Human Diseases | Drug resistance: antineoplastic | 4 |
| Human Diseases | Endocrine and metabolic disease | 5 |
| Human Diseases | Immune disease | 2 |
| Human Diseases | Infectious disease: bacterial | 12 |
| Human Diseases | Infectious disease: parasitic | 2 |
| Human Diseases | Infectious disease: viral | 1 |
| Human Diseases | Neurodegenerative disease | 3 |
注:(1)First Catergory:KEGG代谢通路的7大类:代谢(Metabolism),遗传信息处理(Genetic Information Processing),环境信息处理(Environmental Information Processing),细胞过程(Cellular Processes),生物体系统(Organismal Systems),人类疾病(Human Diseases),药物开发(Drug Development); (2)Second Catergory:KEGG代谢通路的名称; (3)Gene No.:注释到该通路下的基因数量。
3.4 表达量分析
使用软件分别对基因的表达水平进行定量分析,以便后续分析不同样本间基因的差异表达情况,并可通过结合序列功能信息,揭示基因的调控机制。
3.4.1 表达量统计
默认利用表达定量软件RSEM分别对基因和转录本的表达水平进行定量分析,定量指标为TPM,指以转录本的条数为计算单位,使用转录本的条数代替拼接片段数,综合考虑了转录本长度和样品表达的基因数等因素,在一定条件下定量更准,尤其样本间表达基因总数差异很大的时候。
3.4.1.1 表达量分布
表达量分布箱线图

注:横坐标为样本/组别名称,纵坐标为表达量取log10对数处理后的值,图中每种颜色表示一个样本/分组,图示中横线表示样本中基因表达的中位数。
表达量分布小提琴图

注:坐标为样本/组别名称,纵坐标为表达量取log10对数处理后的值,图中每种颜色表示一个样本/分组,图中膨大部分表示整个样本基因表达量最集中的区域。
表达量分布密度图

注:图中横坐标为表达量取log10对数处理后的值,该数值越高,表示基因表达量越高;纵坐标为基因的密度,即为对应横轴表达量的基因数 / 检测已表达基因的总数;图中每种颜色表示一个样本,所有概率的总和为 1,即每个区域的面积均为 1;密度曲线的峰值表示整个样本基因表达量最集中的区域。
3.4.2 样本间表达量分析
根据基因在不同样本间的表达情况,对样本间共有与特有表达基因进行venn分析、相关性和PCA分析。
3.4.2.1 Venn分析
Venn 分析用于展示样本间共有和特有表达的基因。
表达量venn图

注:不同颜色的圆圈代表一组样本表达的基因,其中的数值代表某两组样本或三组样本间共有和特有的表达基因数目,圆内部所有数字之和代表该组表达基因个数的总和,圆的交叉区域代表各组表达基因的共有基因个数(样本数目要求2~6)。
3.4.2.2 相关性分析
生物学重复样本之间的相关性分析,一方面检验生物学重复之间的变异是否符合实验设计的预期,另一方面为差异基因分析提供基本参考。相关系数越接近于1,表明基因在样本间的表达量相似度越高,即样本间相关性越好。
样本间相关性热图

注:图中右侧和下侧为样本名称,左侧和上侧为样本聚类情况,不同颜色的方块代表两个样本的相关性高低。
样本间相关性系数表
| Control_1 | Control_2 | KEGIHAQ_1 | KEGIHAQ_2 | |
|---|---|---|---|---|
| Control_1 | 1 | 0.999 | 0.996 | 0.995 |
| Control_2 | 0.999 | 1 | 0.995 | 0.994 |
| KEGIHAQ_1 | 0.996 | 0.995 | 1 | 0.999 |
| KEGIHAQ_2 | 0.995 | 0.994 | 0.999 | 1 |
注:第一行和第一列为样本名,表中数值为两个样本的相关系数,数值越大,表示两个样本的相关性越大,越相近。
3.4.2.3 PCA分析
主成分分析(PCA)可以降低数据的复杂性,深入挖掘样品之间的关系和变异大小。基本原理是,利用数学的方法,将原来变量重新组合成一组新的互相无关的几个综合变量(即主成分),对所有因素按重要性排序,通常靠后的微小因素被忽略掉,从而起到简化数据的作用。实际项目中,我们可以通过PCA找出离群样品、判别相似性高的样品簇等。
样本间PCA图

注:样本通过降维分析后,在主成分上有相对坐标点,各个样本点的距离代表了样本的距离,距离越近表明样本间相似性越高。横轴表示二维图中主成分1(PC1) 对区分样本的贡献度,纵轴表示二维图中主成分2(PC2) 对区分样本的贡献度。
主成分解释表
| Proportion of Variance | |
|---|---|
| PC1 | 0.429209704 |
| PC2 | 0.341130343 |
| PC3 | 0.229659953 |
注:第一列为各主成分,第二列为主成分所占比例,数值越大代表该主成分越能区分样本。
3.5 表达量差异分析
获得基因的Read Counts数后,可进行样本间基因的差异表达分析,鉴定出样本间差异表达基因,进而研究差异基因的功能。该模块主要对差异基因的进行统计分析,包括差异表达基因详情表(差异基因在单个比较组中的表达情况)和差异表达基因统计表(差异基因在多个比较组别中的表达情况)。
3.5.1 表达量差异分析
基因表达量分析获得基因的Read Counts数后,采用DESeq2、DEGseq或edgeR,可进行样本间或组间基因差异表达进行分析,鉴定出差异表达的基因。
表达量差异统计统计柱状图

注:横坐标代表差异分组,纵坐标代表差异基因的个数,红色为上调基因,绿色为下调基因。
表达量差异统计表
| Gene_id | Gene_name | Gene_description | KEGIHAQ_vs_Control | Sum |
|---|---|---|---|---|
| 3 | 3 | |||
| gene991 | - | DUF2798 domain-containing protein [Lactobacillus delbrueckii] ADY85105.1 Hypothetical protein LBU_0920 [Lactobacillus delbrueckii subsp. bulgaricus 2038] ADY85132.1 Hypothetical protein LBU_0947 [Lactobacillus delbrueckii subsp. bulgaricus 2038] ALT47433.1 membrane protein [Lactobacillus delbrueckii subsp. bulgaricus] ALT47478.1 membrane protein [Lactobacillus delbrueckii subsp. bulgaricus] | yes | 1 |
| gene1786 | - | hypothetical protein LBUL_1819 [Lactobacillus delbrueckii subsp. bulgaricus ATCC BAA-365] ALT48230.1 hypothetical protein AT236_01904 [Lactobacillus delbrueckii subsp. bulgaricus] EHE87972.1 hypothetical protein LDBUL1632_01570 [Lactobacillus delbrueckii subsp. bulgaricus CNCM I-1632] | yes | 1 |
| gene1785 | endA | DNA-entry nuclease [Lactobacillus delbrueckii subsp. bulgaricus] | yes | 1 |
注:(1)Gene_id:基因的ID编号;(2)Gene name:基因名称;(3)Gene description:基因描述信息;(4)其余几列为该比较组中识别到的差异基因数目; (5)Sum:为该基因显著差异表达的次数;(6)Gen Type:表示已知基因ref gene或新基因novel gene或sRNA;
表达量差异统计统计堆积图

注:横坐标代表差异分组,纵坐标代表差异基因的个数,红色为上调基因,绿色为下调基因。
表达量差异散点图

注:横纵坐标分别表示两组样本中基因的表达量,这里横纵坐标的数值都做了对数化处理,每个点代表一个特定的基因。特定的一个点对应的横坐标值为该基因在对照组中的表达量,纵坐标值为该基因在处理组中的表达量。图中红色点表示显著上调的基因,绿色点表示显著下调的基因,灰色点为非显著差异基因。将所有基因映射上去后,越接近0的点,说明表达量越低;那些偏离了对角线程度越大的点表明该基因在两个样本间表达差异越大。
表达量差异火山图

注:横坐标为基因在两组样本间表达差异的倍数变化值,即FC值。纵坐标为基因表达量变化差异的统计学检验值,即p值。p值越高则表达差异越显著,横纵坐标的数值都做了对数化处理。图中每个点代表一个特定的基因,红色点表示显著上调的基因,绿色点表示显著下调的基因,灰色点为非显著差异基因。将所有基因映射上去之后,可以获知,在左边的点为表达差异下调的基因,右边的点为表达差异上调的基因,越靠两边和上边的点表达差异越显著。
3.6 基因集分析
基因集, 即根据一定的筛选条件(比如功能、 表达量以及表达差异情况等),获得的基因集( gene list)。其目的是从众多基因中挖掘与研究目的或表型相关的一些基因,并进行功能、表达等研究。
3.6.1 功能注释分析
3.6.1.1 COG注释
COG 是Clusters of Orthologous Groups of proteins的缩写(http://www.ncbi.nlm.nih.gov/COG/)。COG是在对已完成基因组测序的物种的蛋白质序列进行相互比较的基础上构建的,COG数据库选取的物种包括各个主要的系统进化谱系。每个COG家族至少由来自3个系统进化谱系的物种的蛋白所组成,所以一个COG对应于一个古老的保守结构域。构成每个COG的蛋白被假定来自于同一个祖先蛋白。进行COG数据库比对可以对预测蛋白进行功能注释、归类以及蛋白进化分析。
基因集COG分类统计柱状图

注:横坐标代表一个COG的功能类型(用大写字母A~Z表示,具体含义见COG注释分类统计表所示);纵坐标表示具有该类功能的基因数目。当选择基因集有两个时,“切换图形”可选择簇状柱形图和双向柱形图,其中双向柱形图中0坐标点上下两侧表示两个分组;当选择基因集有三个及以上时,“切换图形”只能选择簇状柱形图。
3.6.2 功能富集分析
对基因集中基因进行功能富集分析,包括GO富集和KEGG富集。
采用软件 Goatools 对基因集中的基因进行GO富集分析,从而获得该基因集中的基因主要具有哪些GO功能。使用方法为 Fisher 精确检验,当经过校正的 P 值(Padjust)<0.05时,认为此GO功能存在显著富集情况。
采用R脚本对基因集中的基因进行KEGG PATHWAY富集分析, 计算原理同GO功能富集分析,当经过校正的 P 值(Padjust)<0.05时,认为此KEGG PATHWAY功能存在显著富集情况。
3.6.2.1 多基因集富集气泡图
3.6.3 Ipath分析
利用iPath2.0 (http://pathways.embl.de)对代谢途径进行可视化分析,可以查看整个生物系统的代谢通路信息。图中的节点代表不同的化合物,边界代表不同的酶促反应。iPath2.0 能够概述次生代谢物的生物合成和重要的调控途径,轻松查找复杂的代谢通路。
iPath代谢通路图

注:图片代表基因集注释的通路,桃红色和浅绿色分别代表不同基因集中的基因注释的通路,蓝色代表两个基因集中的基因共同注释的通路。其中,Map:Metabolic pathways 代表代谢通路;Regulatory pathways代表调控通路;Biosynthesis of secondary metabolites代表次级代谢产物合成。
3.7 sRNA分析
细菌中,长度在50~500nt的ncRNA通常定义为小RNA(small RNA, sRNA),它们主要位于基因间区,也有的位于编码基因5’和3’UTR区域。研究表明,这类sRNA参与很多生物学过程当中,如细菌的转录调控、RNA加工与修饰、mRNA稳定、mRNA翻译、蛋白质降解、质粒复制和细菌感染等,在细菌适应不同的自然环境中发挥重要的调控作用。
3.7.1 sRNA预测
sRNA预测根据当前主流的原核转录组分析软件Rockhopper(http://cs.wellesley.edu/~btjaden/Rockhopper/)获得sRNA预测结果,预测依据主要是碱基测序覆盖度信息。
3.7.2 sRNA注释
利用Blast及公共数据库sRNAMap,sRNATarBase,SIPHT及Rfam资源对鉴定到的sRNA进行注释,注释的统计结果使用Venn图可视化。
3.7.2.1 Rfam注释
Rfam注释统计图

注:不同颜色的圆圈代表Rfam数据库中不同的RNA类型。
Rfam注释统计表
| Type | Total Number | Total Percent (%) |
|---|---|---|
| sRNA | 1 | 16.67 |
| Gene | 3 | 50 |
注:(1)Type :RNA类型;(2)Total Number:不同RNA类型的数目;(3)Total Percent (%):不同RNA类型占总RNA的百分比。
3.8 转录本结构分析
转录本结构分析包括操纵子分析、转录起始和终止位点预测、UTR注释及分析、启动子序列预测和SNP/InDel分析。
3.8.1 操纵子分析
在原核生物的基因组中很多功能相关的基因前后相连成串,由一个共同的控制区进行转录调控,这样的基因结构称为操纵子(operon)。操纵子一般由启动基因、操纵基因和一系列紧密连锁的功能基因组成。
3.8.2 UTR注释及分析
UTR(Untranslated Regions)即非翻译区,是mRNA分子两端的非编码片段,包括5’UTR与3’UTR,其中,5’UTR指的是从转录起始位点至起始密码子的一段非翻译区。3’UTR指的是从编码区末端的终止密码子至终止子的一段非翻译区。其在mRNA的稳定性及翻译等生物学过程中发挥着重要的调控作用。
UTR长度分布图

注:图中横坐标为UTR Length,该数值越高,表示UTR越长;纵坐标为UTR频率,即为对应横轴长度的UTR总数。
UTR长度分布表
| UTR Length (bp) | Count |
|---|---|
| 1~50 | 1653 |
| 51~100 | 177 |
| 101~150 | 68 |
| 151~200 | 20 |
| 201~250 | 24 |
| 251~300 | 20 |
| 301~350 | 13 |
| 351~400 | 1 |
| 401~450 | 3 |
| 451~500 | 1 |
| 501~550 | 7 |
| 551~600 | 1 |
| 601~650 | 2 |
| 651~700 | 2 |
| 701~750 | 0 |
| 751~800 | 2 |
| 801~850 | 4 |
| 851~900 | 2 |
| 901~950 | 5 |
| 951~1000 | 1 |
| >1000 | 19 |
| Totle | 2025 |
注:(1)UTR Length :UTR长度;(2)Count:对应长度的UTR总数。
3.8.3 SNP/InDel分析
SNP全称Single Nucleotide Polymorphisms,是指在基因组上由单个核苷酸变异形成的遗传标记,其数量很多,多态性丰富。SNP在CG序列上出现最为频繁,而且多是C转换为T,原因是CG中的C常为甲基化的,自发地脱氨后即成为胸腺嘧啶。根据SNP特点分为转换和颠换。InDel(insertion-deletion)是指相对于参考基因组,样本中发生的小片段的插入缺失,该插入缺失可能含一个或多个碱基。
3.8.3.1 SNP/InDel变异位点注释统计
变异位点注释统计饼图

注:图中不同的颜色代表参考基因组的不同区域,扇形的面积越大表示该区域中包含的SNP/InDel数目越多。
SNP Indel变异位点注释统计表
| Region | Control_1 | Control_2 | KEGIHAQ_1 | KEGIHAQ_2 |
|---|---|---|---|---|
| downstream | 8 | 4 | 8 | 6 |
| exonic | 180 | 154 | 171 | 144 |
| upstream | 11 | 13 | 10 | 13 |
| upstream;downstream | 37 | 41 | 37 | 28 |
| total | 236 | 212 | 226 | 191 |
注:统计不同区域中SNP/InDel数目分布情况。 (1) exonic:外显子区域;(2) intronic:内含子区域;(3) intergenic:基因间区域;(4) upstream:转录起始位点上游1000bp以内的区域;(5) downstream:转录终止位点下游1000bp以内的区域;(6) upstream;downstream:上个基因转录起始位点下游1000bp以内的区域同时位于下个基因上游1000bp以内的区域;(7) UTR5:5’端非翻译区域;(8) UTR3:3’端非翻译区域;(9) UTR5;UTR3:在上个基因3’端非翻译区域以及下个基因5’端非翻译区域;(10) ncRNA:非编码RNA区域。
3.8.3.2 SNP统计
SNP类型统计柱状图

注:横坐标表示SNP的类型,纵坐标表示该SNP类型在不同样本中的存在数目。
SNP类型统计饼图

注:不同SNP类型在各样本中的存在数目统计结果。
SNP类型统计表
| Type | Control_1 | Control_2 | KEGIHAQ_1 | KEGIHAQ_2 |
|---|---|---|---|---|
| Transition | ||||
| T/C | 54 | 47 | 48 | 52 |
| G/A | 31 | 27 | 39 | 23 |
| A/G | 53 | 58 | 47 | 46 |
| Transversion | ||||
| A/T | 5 | 6 | 4 | 2 |
| G/T | 13 | 7 | 11 | 7 |
| T/A | 9 | 7 | 5 | 11 |
| G/C | 7 | 5 | 4 | 4 |
| C/G | 4 | 4 | 7 | 1 |
| C/A | 13 | 11 | 10 | 6 |
| A/C | 5 | 9 | 9 | 9 |
| total | 194 | 181 | 184 | 161 |
注:第一列Type表示SNP 类型,包括Transition(转换,由一种嘧啶变成另一种嘧啶,或一种嘌呤变成另一种嘌呤)和Transversion(颠换,嘌呤与嘧啶之间的交换),其他列表示该SNP类型在不同样本中的存在数量。
四、备注
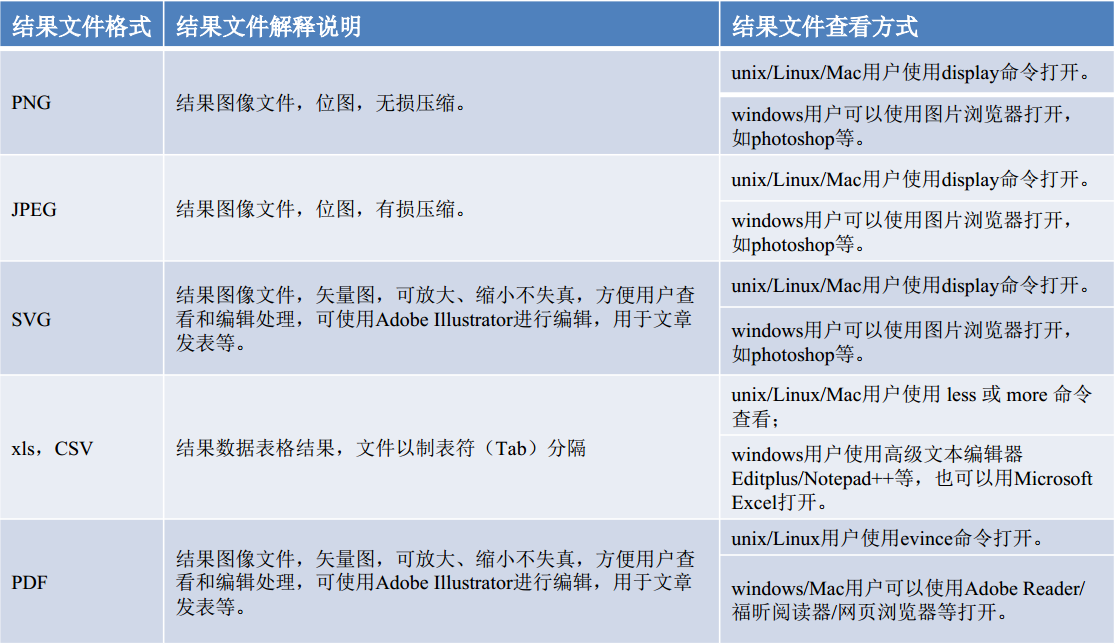
4.1 结果文件查看说明